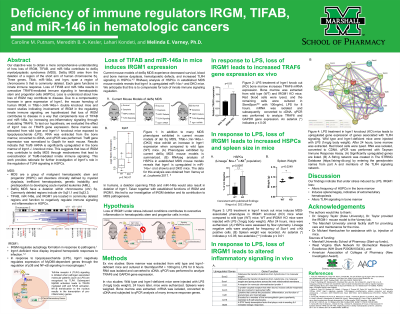
Objective : Our objective was to obtain more comprehensive understanding of how loss of IRGM, TIFAB, and miR-146a contribute to del5q myelodysplastic syndromes (MDS). Del5q MDS arise from the deletion of a region of the short arm of human chromosome 5q. Three genes, Tifab, miR-146a, and Irgm, span a region of chromosome 5 that is commonly deleted. Each gene functions in innate immune response. Loss of TIFAB and miR-146a results in overactive TRAF6-mediated immune signaling in hematopoietic stem and progenitor cells (HSPCs). Less is understood about how loss of IRGM may contribute to disease. Due to a compensatory increase in Irgm1, the mouse homolog of human IRGM, gene expression in Tifab-/-;miR-146a-/- double knockout mice and recent studies indicating involvement of IRGM in the regulating innate immune signaling, we hypothesized that loss of IRGM contributes to disease in a way that complements loss of TIFAB and miR-146a, by increasing pro-inflammatory signaling through modulating TRAF6.
Methods: To test our hypothesis, we evaluated the effect of Irgm1 loss on TRAF6 gene expression. Bone marrow was extracted from wild type and Irgm1-/- knockout mice. RNA was extracted from the bone marrow, converted to cDNA, and qPCR was performed. Traf6 gene expression was normalized to Gapdh for each mouse.
Results: Results indicate that Traf6 mRNA is significantly upregulated in the bone marrow of Irgm1-/- knockout mice.
Conclusions: This suggests that IRGM may function in suppressing TRAF6 gene expression and that loss of IRGM may contribute to del5q MDS through mechanisms that lead to overactivation of Traf6-mediated innate immune signaling. This work provides rationale for further investigation of Irgm1’s role in the regulation of TLR4 signaling in HSPCs.